Entdecker der verborgenen Metabolite
von Prof. Dr. Sebastian Böcker
Alles, was lebt, hat Metabolite, produziert Metabolite und verbraucht Metabolite. Die chemischen Prozesse zur „Verarbeitung“ dieser Moleküle sind gemeinhin als Stoffwechsel bekannt. Damit haben sie nicht nur eine enorme Bedeutung für unser Leben, sondern sie liefern auch wertvolle Informationen über den Zustand eines Lebewesens oder einer Umgebung. Doch die Diversität dieser chemischen Verbindungen bereitet der Wissenschaft einige Schwierigkeiten. Nur vergleichsweise wenige Moleküle wurden bisher in ihrer Struktur aufgeklärt, und wir wissen bislang nicht einmal, was wir nicht wissen – wie viele Metabolite also noch auf die Aufklärung ihrer Struktur warten. Die praktische Bedeutung dieser Strukturaufklärung lässt sich am Beispiel von Wirkstoffen aus der Natur zeigen: Diese sind entzündungshemmend, können Krankheitskeime abtöten, oder unterbinden das Wachstum von Krebszellen. Weit mehr als ein Drittel aller heute verfügbaren Medikamente basieren auf solchen sekundären Naturstoffen, wie sie in zahlreichen Pflanzen, Bakterien und Pilzen vorkommen. Den reichhaltigen Medizinschrank der Natur nutzbar zu machen und neue Naturstoffe zu identifizieren, ist jedoch zeit-, kosten- und arbeitsintensiv.
Weil Naturstoffe und andere Metabolite üblicherweise in extrem niedrigen Konzentrationen vorkommen – oft sind weniger als ein Millionstel Millionstel Millionstel Gramm in einer Probe – nutzt die Wissenschaft Massenspektrometrie, um diese Moleküle nachzuweisen. In aller Regel werden dabei aber nur die Moleküle identifiziert, die durch den Abgleich mit einer Datenbank von Referenzmessungen, also kommerziell erhältlichen Reinsubstanzen, eindeutig zugeordnet werden können. Auf diese Weise lässt sich leider niemals die Struktur eines gänzlich „neuen“ Moleküls aufklären. (Natürlich ist das Molekül nicht wirklich neu, die Natur produziert es ja schon seit Millionen von Jahren; nur der Menschheit ist seine Struktur noch gänzlich unbekannt.)
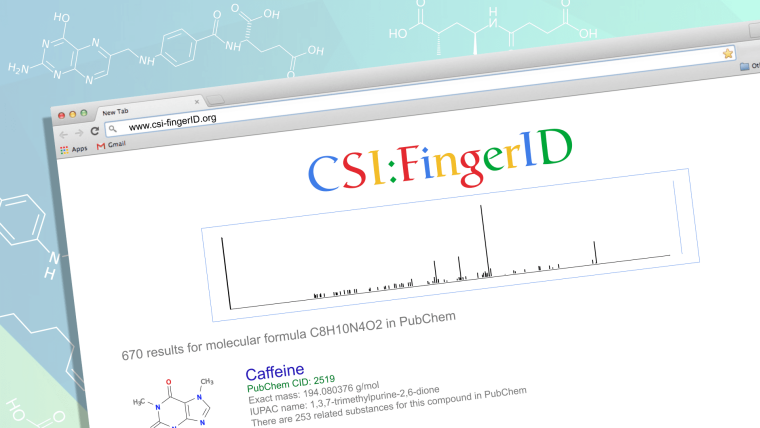
Hier kommt die Bioinformatik ins Spiel: Wir entwickeln beispielsweise Methoden, die es erlauben, mit den Massenspektrometriedaten in einer Molekülstruktur-Datenbank zu suchen. Unsere Suchmaschine für Molekülstrukturen – CSI:FingerID – kann man sich vorstellen wie eine Suche im Internet: Zu einer Anfrage (einem gemessenen Massenspektrum) liefert sie eine geordnete Liste von Treffern (Molekülstrukturen); im besten Fall findet sich die korrekte Antwort an der ersten Stelle. Anders als bisher muss dazu lediglich die Struktur eines Moleküls hinterlegt werden, aber die Substanz nicht kommerziell erhältlich sein. Das funktioniert auch dann, wenn diese Moleküle noch nie beobachtet wurden: So haben wir beispielsweise die Strukturen von mehr als 28.000 theoretisch möglichen Gallensäuren generiert.

Worfklow und Interface der SIRIUS Software
Abbildung: Sebastian Böcker, SIRIUS Nutzerinterface